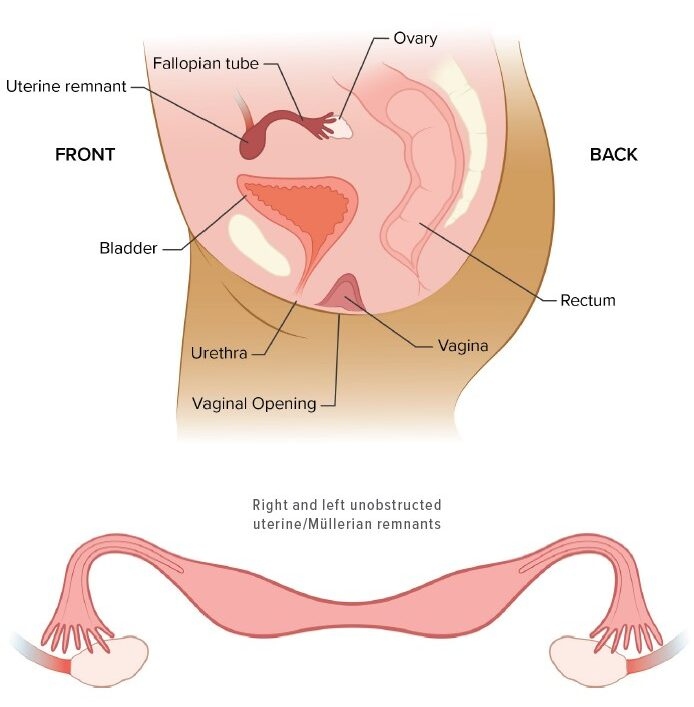
- Vaginal and Uterine Agenesis: MRKH is a congenital condition where the vagina and sometimes the uterus are underdeveloped or absent, though external genitalia and secondary sexual characteristics are typically normal.
- Symptoms: Primary amenorrhea (absence of menstruation) by age 16, and a short or absent vaginal canal are common signs.
- Diagnosis: Diagnosed through physical exams, imaging (ultrasound or MRI), and sometimes genetic testing to confirm the condition and assess associated anomalies.
- Treatment: Vaginal reconstruction (dilation or surgery) and fertility options (such as surrogacy or egg donation) help address sexual function and infertility, with psychosocial support for emotional well-being.
Vaginal Agenesis/Mayer-Rokitansky-Küster-Hauser (MRKH) Syndrome: Detailed Overview
Vaginal agenesis, also known as Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome, is a congenital condition where a woman is born without a fully developed vagina and, in many cases, a malformed or absent uterus. This condition typically does not affect the external genitalia, and individuals with MRKH often have normal secondary sexual characteristics, such as breast development and pubic hair growth, because their ovaries usually function normally.
The condition is typically diagnosed in adolescence when a female patient does not begin menstruating and lacks a fully formed vagina. MRKH can affect fertility and sexual function but is not life-threatening.
What is MRKH Syndrome?
MRKH syndrome is a rare condition that occurs in approximately 1 in 4,500 to 5,000 live female births. It primarily affects the reproductive system, and while it is present from birth, it may not be recognized until puberty, when menstruation does not occur, or during a routine pelvic examination.
The disorder is classified into two types:
- Type 1 (Classic MRKH): Involves vaginal agenesis with absent or underdeveloped uterus but normal ovaries. External genitalia (vulva) typically appear normal, and secondary sexual characteristics develop normally.
- Type 2 (MRKH with associated anomalies): This type includes vaginal agenesis, absent uterus, and associated renal, skeletal, or cardiac anomalies. It is less common and can be more complex.
Causes of MRKH Syndrome
The exact cause of MRKH is not fully understood, but it is believed to be a genetic disorder, likely involving developmental disruptions during embryogenesis (fetal development). The condition is associated with problems during the formation of the Müllerian ducts, which are responsible for the development of the uterus, fallopian tubes, and vagina. Some possible causes and contributing factors include:
- Genetic Mutations: MRKH may be linked to specific mutations, although a clear inheritance pattern is not always observed. Certain genetic syndromes or deletions have been implicated in MRKH, including WNT4, LHX1, and others.
- Environmental Factors: Environmental exposures during pregnancy, such as certain medications or endocrine disruptors, may increase the risk of developmental issues.
- Sporadic Occurrence: Many cases of MRKH syndrome occur without any identifiable family history, making it difficult to predict or prevent.
Symptoms of MRKH Syndrome
The most common symptoms of MRKH are related to the reproductive system:
- Absence of Menstruation (Primary Amenorrhea): The most notable symptom is the absence of menstruation by the age of 16, despite normal breast development and pubic hair growth.
- Vaginal Agenesis: Affected individuals may have a very short, underdeveloped vaginal canal, or a completely absent vagina. This results in an inability to have penetrative intercourse.
- Normal External Genitalia: The vulva (external genitalia) is typically normal, and the development of secondary sexual characteristics (e.g., breast development, pubic hair) is usually unaffected.
- Absence or Abnormal Uterus: The uterus may be entirely absent, or it may be underdeveloped or malformed. In some cases, individuals may have a rudimentary uterus, which cannot support pregnancy.
- Associated Anomalies (Type 2): In cases of MRKH type 2, renal abnormalities, such as absent or malformed kidneys, and skeletal issues, such as spinal deformities, may also be present.
Diagnosis of MRKH Syndrome
The diagnosis of MRKH is typically made during adolescence, after the onset of puberty when menstruation does not occur. Diagnostic steps include:
- Physical Examination: The external genitalia are usually normal, and a pelvic examination can reveal the absence of a developed vagina.
- Ultrasound or MRI: Imaging tests, such as pelvic ultrasound or MRI, help confirm the presence or absence of the uterus and assess other abnormalities. An MRI is particularly useful in visualizing the vaginal cavity, uterus, and surrounding organs.
- Genetic Testing: Genetic testing can help identify any underlying genetic abnormalities, particularly in cases where associated anomalies are present (type 2 MRKH).
- Karyotype Analysis: In most cases, individuals with MRKH have a normal female karyotype (XX), meaning they have two X chromosomes, which is characteristic of females.
Treatment Options for MRKH Syndrome
While MRKH is a congenital condition with no cure, there are treatment options to address the symptoms and enhance quality of life. Treatment may include:
1. Vaginal Reconstruction
- Vaginal Dilation: The most common method for creating a functional vaginal canal. The patient uses vaginal dilators (a set of graduated plastic tubes) to gradually expand the vaginal tissue and create a functioning vaginal opening. This process requires patience and regular maintenance but can allow for normal sexual function.
- Surgical Options: If dilation is not successful or preferred, there are surgical procedures, such as the McIndoe procedure, which uses skin grafts or other tissues to construct a functional vagina. Vaginal reconstructive surgery is a more invasive option, often used when dilation is not feasible.
2. Fertility Treatments
- Surrogate Pregnancy: Since women with MRKH typically lack a uterus, they are unable to carry a pregnancy. However, if the ovaries are functional and producing eggs, surrogacy is an option. The woman’s eggs may be fertilized using in vitro fertilization (IVF), and the embryo is implanted into a surrogate’s uterus.
- Egg Donation: In some cases, women may need to use donor eggs if their ovaries are not functional, but this depends on the specific diagnosis.
3. Psychosocial Support
- Coping with the diagnosis of MRKH can be challenging, particularly for adolescents. Psychological counseling and support groups are valuable resources to help individuals and families navigate the emotional and social aspects of the condition.
4. Management of Associated Anomalies
- If the individual has associated abnormalities such as renal or skeletal defects, those will require specialized treatment. For example, kidney abnormalities may require nephrologic evaluation and monitoring.
Long-Term Outlook for Individuals with MRKH
- Normal Life Expectancy: Individuals with MRKH typically have a normal life expectancy, as the condition is not life-threatening.
- Sexual Function: Vaginal dilation and/or reconstructive surgery can help restore sexual function, though this requires ongoing maintenance.
- Fertility: Although affected women cannot carry a pregnancy, many have the option of having biological children through surrogacy or egg donation.
- Emotional Well-Being: With the right support, most individuals can lead fulfilling lives. Psychological support is important to address any feelings of distress or social concerns.
Conclusion
Vaginal agenesis/Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome is a congenital condition that affects the development of the vagina and uterus, leading to primary amenorrhea (lack of menstruation) and possible infertility. However, individuals with MRKH typically have normal external genitalia and normal secondary sexual characteristics. Early diagnosis, support, and appropriate treatment options like vaginal dilation, reconstructive surgery, and fertility assistance can help affected women lead fulfilling lives.
